
Table of Contents
- Preparation of Anhydrous Cerous Fluoride
- Fluoride Electrolytes
- Electrowinning
- Laboratory-Cell Development
- Cell Type No. 1
- Preparation of Charge
- Melting of Charge
- Electrolytic Operations
- Cell Type No. 2
- Cell Type No. 3
- Cell Type No. 4
- Reno Cerium Electrowinning Cell (Cell Type No. 5)
- Electrowinning Cell and Electrode Assembly
- Steel Glove Box
- Replacing Air with Inert Atmosphere
- Melting of Bath
- Cerium Electrowinning Run (CE-42)
- Analyses of Cell-Box Gases
- Lithiothermic Reduction and Vacuum Refining
- Ceramics
- Electrical Conductivity of Cerium Metals
The metallurgical laboratory data developed in this study indicate an improved electrowinning process for producing higher purity cerium ingot. Equipment under development included the molten-bath container or electrolytic cell, electrodes, and related items, as well as an enclosure or cell box resembling that of a vacuum furnace. The cell box is designed to allow molten electrolysis to be performed in an atmosphere that can be closely controlled as to composition, temperature, and pressure. The molten electrolysis was conducted in a graphite cell, in which the temperature was also under close control and which prevented contact between cerium and carbon and permitted deposition of cerium metal at a temperature as close as possible to 805° C. Cell off-gases were sampled and analyzed. A section of this report describes these procedures and discusses the relation of cell-gas composition to metal purity.
In an experimental study of the suitability of several mixtures of metallic halides as the solvent phase of molten-salt electrolytes, the mixture of cerium, barium, and lithium fluorides in the proportion of 73, 12, and 15 percent, respectively, showed the greatest promise. Nevertheless, studies on the compounding and properties of cerium electrolytes are being continued, as the electrolyte is the cornerstone of the process.
Electrocerium nodules were formed in the small cells described in this report. These irregular, rounded nodules resulted from cerium metal being deposited in a molten condition and slipping down the cathode to the end where they sank into the bath and solidified. However, it is believed that in a larger cell and under favorable conditions the nodules will coalesce.
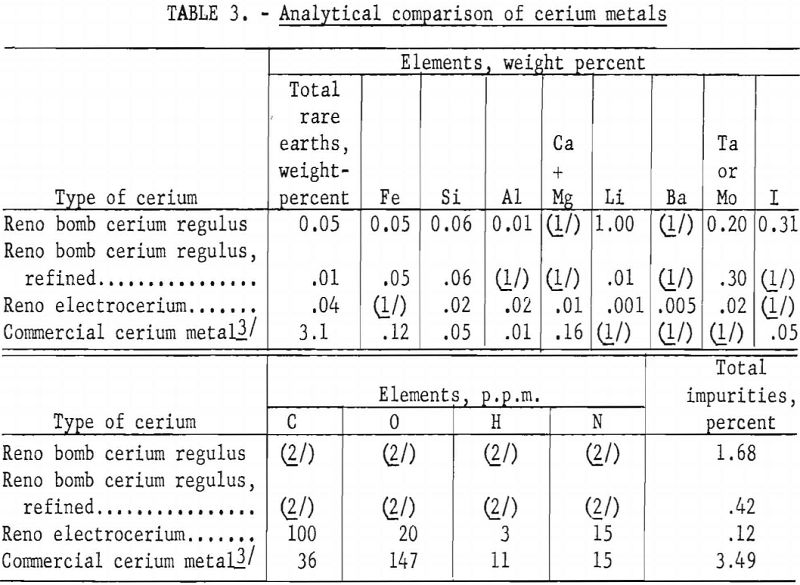
One of the interesting phenomena observed, which is still under investigation in this higher purity cerium electrowinning laboratory, is the large variation in purity of different nodules of metal from the same cathode in the same cell run. For example, although 0.01 percent calcium plus magnesium is typical in electrocerium analysis, a number of nodules of cerium have been made in which no magnesium or calcium has been detected. A method of comparing the nodules by air corrosion was used to classify higher- from lower-grade metal.
As an indicator of metal purity and an aid to learning more about certain properties of cerium, electrical conductivity measurements of the metal over a span of temperatures in a low range have yielded valuable data. Equipment for measuring electrical conductivity, like the electrowinning equipment, is still in a developmental stage. However, data obtained to date have shown a distinct correlation between electrical conductivity and metal purity. For example, at 0° C. the conductivity of a nodule of Reno electrocerium 99.9 percent pure was more than three times that of a sample of 95.8-percent cerium metal obtained from a commercial source.
A laboratory lithiothermic reduction procedure for preparing cerium regulus on a 50-gram scale by lithium-iodine reduction of cerous fluoride and the vacuum refining of this regulus to cerium metal are also described.
The growth and future importance of the rare-earth metals in commercial industry may depend largely upon improved methods of extracting and refining these elements. It is proposed to use the data on the reduction and refining of cerium given in this report for this objective.
Cerium is one of the elements in a group comprising 65 percent of the known metallic elements that have never been prepared in sufficient quantity and quality to allow development of their important use patterns; more than 80 percent of all known elements are metallic, and only 35 percent of the metallic elements are being applied commercially to any major extent.
Cerium is the most abundant element of the rare-earth group. Mine-run ores of cerium from Mountain Pass, Calif., contain 3.6 percent cerium; monazite minerals from Florida beach sands, South Carolina, India, and Brazil range from 25 to 26 percent cerium.
Cerium is a metallic element with a silvery luster, atomic weight of 140.13, density of 6.9, and atomic volume of 20, which forms crystals of face-centered cubic and close-packed hexagonal structures; it has a melting point of 804° ±5° C., according to F. H. Spedding.
Because of its superreactive nature, cerium alloys or forms compounds with virtually all metallic, nonmetallic, and gaseous elements. Felix Trombe reports that cerium metal has distinctive elemental or metallic properties, such as its volume abnormalities, either under pressure or at low temperatures. In a study of metallic cerium in 1934, a nonreversible magnetic cycle was identified that did not seem to depend on the quantity of iron (of the order of 100 p.p.m.) in the cerium. These data have been confirmed after some years by Charlotte Henry La Blanchetais who succeeded in preparing cerium with less than 5 p.p.m. of iron. This higher purity metal follows the Curie-Weiss law; that is, the reciprocal of the magnetic susceptibility is a straightline function of the absolute temperature. However, with 100 p.p.m. iron, the temperature-susceptibility relationship was altered markedly, showing that the magnetic properties of cerium are very sensitive to traces of iron. Other scientists did not confirm this phenomena, and the possible influence of iron was debated. In 1945 Trombe and Foex presented new data showing that the magnetic cycle is accompanied by a dilatation cycle of considerable importance, corresponding to a contraction in volume at diminishing temperature and an expansion in volume of 10 percent at increasing temperature. Instead of the 10-percent contraction at 109° K., there is a certain inertia and expansion around 175° K., then the two curves rejoin.
As the metal already has a dense structure (face-centered cubic) at ordinary temperatures and a contraction of 10 percent at lower temperatures, Linius Pauling presented the idea that this phenomenon is electronic; that is, he considered that an electron changes orbit at lower temperatures, which caused contraction of cerium’s atomic radius. Actually, cerium is again face-centered cubic at lower temperatures but has different parameters.
If pure cerium is submitted to successive heatings and coolings within a certain temperature range, the importance of magnetic and dilatation cycles diminishes progressively until their suppression is nearly complete. Cerium is then changed into another form, likewise compact but hexagonal.
Felix Trombe and Marc Foex have established that certain impurities are responsible for the absence of cycles, regardless of the thermal treatment of the metal. It has been reported that calcium and magnesium, especially,are deterrents to obtaining the contracted form of cerium. In 1949 Lawson and Tang obtained a transformation at 15,000 kg. per sq. cm. corresponding to a decrease in volume of 16.5 rather than 10 percent. This transformation was produced under pressure at ordinary temperature, whereas under standard pressure a temperature of 109° K. would be required.
These observations, reported by Trombe and other investigators, illustrate the importance of purity on certain properties of metallic cerium. Even the minute amount of contaminants in the purest cerium that has been studied may camouflage some as-yet undiscovered properties of still purer metal. It is possible that one or more of the elusive properties of cerium will lead to important new uses. Another approach to greater utilization of this abundant but little-used natural resource is to find less expensive ways of producing a marketable-grade product. The aim of the Bureau’s research is to develop processes that will yield high-purity metal that has commercial potentialities. Refining techniques for producing ultrapure metal, such as single-crystal or “whisker” products, are beyond the scope of present research at Reno but will become an essential phase of reduction and refining work in the near future.
Since the work reported by F. Trombe in 1956 and 1957, the Bureau’s reduction and refining laboratory at Reno has reported the presence of molybdenum and tantalum in cerium metal where these metals are held in contact above 820° C. for any appreciable time.
Cerium metal has been prepared by calcium reduction and has been electrowon from chloride and fluoride electrolytes. W. J. Kroll states:
Fluorine metallurgy is in its infancy, and it is much less advanced than that of chlorine. The advent of large fluorine cells has brought about considerable advance in organic fluorine compounds, and it is hoped that this movement will make itself felt also in the field of inorganic chemistry and its application to metallurgy.
The electrolysis of aqueous solutions of inorganic salts of cerium does not deposit pure metal, because the nascent cerium atoms react with water or hydrogen to form metallic oxide and hydrated oxide, or cerium hydride.
Electrolysis of alcoholic solutions of the anhydrous chlorides of cerium has been investigated.
Recent literature describes the hydrogen reduction of CeO2 to Ce2O3 and nonstoichiometric intermediate oxides at temperatures between 250° and 1,400° C. The extent of reduction increases with the temperature.
The Reno process for electrowinning cerium from CeO2 in fluoride electrolytes is similar to electrowinning aluminum from Al2O3 in cryolite baths, except for the more reactive nature of molten cerium. The Reno type of cerium electrowinning cell, with its controlled temperature and atmosphere, was a natural laboratory development which was partly suggested by previous work on the inert-atmosphere, electric-arc welding of tantalum and molybdenum cans for lithium-bomb reduction of cerous fluoride. In the lithiothermic work it was found that both tantalum and molybdenum alloy with cerium metal at the temperatures, pressures, and other conditions encountered in bomb reductions. The analysis of cerium metal from the tests showed tantalum 0.3 percent and molybdenum 2.7 percent.
The development of the 6-inch diameter, laboratory graphite cell was the result of investigations which proved that graphite was an excellent refractory or corrosion-resistant material with molten rare-earth fluorides and oxyfluorides under controlled temperature-and-atmosphere electrolytic conditions. Graphite also costs much less than any suitable metallic container, can be easily fabricated into any size or shape, and is stable at 900° C. in the cell atmosphere consisting of argon or helium with CO2, CO, and a small amount of O2.
The following basic data guided the selection and composition of the CeF3-BaF2-LiF solvent-phase electrolyte for electrowinning of cerium from CeO2:
- The electrolyte has a melting point 75° to 80° C. below that of cerium.
- The fluorides were chosen instead of the chlorides because they are less hygroscopic. Hydrogen forms cerium hydrides with molten cerium.
- Sodium and potassium fluorides were found to be electrolytically unstable at the temperatures and pressures used in the cell.
- The relation of density of cerium metal at 6.9 and the oxyfluoride electrolyte at 4.3 was advantageous.
- Electrical resistivities of the molten bath were in the proper range for temperature control, electrolytic oxidation-reduction reactions, or transfer of cerium cations and oxyfluoride anions.
When these investigations on rare metals were initiated in 1956 at Reno, Nev., considerable metallothermic effort had been devoted to the preparation of rare-earth metals at Ames, Iowa, by F. H. Spedding and his coworkers and by the Bureau of Mines station at Albany, Oreg. Therefore, the metallothermic work at Reno was limited to lithium-iodine reductants. This phase of the reduction and refining laboratory work was completed in fiscal year 1958, and the data are given in this report.
Preparation of Anhydrous Cerous Fluoride
Anhydrous cerous fluoride was made for the preparation of cerium metal by lithiothermic reduction and for an electrowinning bath constituent.
Anhydrous cerous fluoride was made by the reaction of ammonium bifluoride with ceric oxide at 500° to 600° C. in air. Attempts to make cerous fluoride at temperatures lower than 500° C. resulted in contamination with ceric oxide. When temperatures exceeded 600° C. cerium oxyfluoride was formed. Platinum dishes or sintered cerous fluoride boats were used as reaction containers. When porcelain, iron, nickel, and fused alumina containers were used, the cerous fluoride product contained silicon, iron, nickel, and aluminum.
Two other methods of preparing anhydrous rare-earth fluorides are described in the literature. One method is precipitation from aqueous solution with hydrofluoric acid and dehydration. The principal difficulties are in the mechanics of washing and drying the hydrated fluoride to prepare anhydrous cerous fluoride consistently free from oxyfluorides. Another method is the gaseous fluorination of ceric oxide with HF or ClF3. According to Von Wartenberg, CeF3 was made by passing anhydrous HF over CeO2 in a platinum tube at temperatures above 400° C. Popov and Knudson describe the fluorination of the oxides lanthanum to samarium with ClF3.
The ammonium-bifluoride method used at Reno does not require the handling of a gelatinous precipitate and is simpler than either of the above methods for preparing small laboratory batches of anhydrous cerous fluoride.
Details of Reno Ammonium Bifluoride Method
Ceric oxide, obtained from a commercial company or from other work at the Reno station, was pulverized in a porcelain mortar with approximately 10 percent excess of the stoichiometric quantity of commercial ammonium bifluoride flakes. The mixture was placed in a platinum dish or CeF3 boat in a laboratory muffle open to the air. The muffle was maintained at 500° to 600° C. for 2 hours before the product was removed. Some ammonium fluoride collected on the inside of the muffle.
The purity of cerous fluoride batches was determined by the X-ray and spectrographic analytical laboratories. A typical spectrographic analysis indicated that aluminum, calcium, iron, lithium, magnesium, nickel, platinum, silicon, and zinc were not detected. An X-ray diffraction pattern showed CeF3. No ceric oxide or cerium oxyfluoride was detected. A sample of the CeF3 was reported by the Bureau of Mines Boulder City station to contain 0.004 percent nitrogen.
One-hundred-and-fifty grams of cerous fluoride made by this method was sent to K. K. Kelly, Chief, Berkeley Thermodynamics Research Laboratory, at Berkeley, Calif, Analyses in the Berkeley station laboratory showed:
In order to prepare larger batches of cerous fluoride without contamination and to eliminate the use of platinum ware, sintered cerous fluoride boats were developed. Cerous fluoride, prepared in platinum dishes as described, was mixed with Carbowax and pressed at 56,000 p.s.i. into 1-13/16- inch, i.d. cylinders. The cylinders were cut in half longitudinally and the resulting boats placed in a 2-9/16-inch i.d. vitrified alumina tube within a horizontal tube furnace. The boats were heated in air at 250° C. for 2 hours to remove the Carbowax and sintered by bringing them to 530° C. in 15 hours.
The sintered cerous fluoride boats were loaded with the ammonium bifluoride-ceric oxide mixture (proportions as previously given), and the tube furnace was brought from room temperature to 500° C. in 4 hours. The charge was maintained at 500° to 600° C. in the air for 2 hours. Eight-hundred-gram batches of cerous fluoride were made by this method.
In a typical spectrographic analysis no aluminum, calcium, iron, lithium, magnesium, nickel, platinum, silicon, and zinc were detected. X-ray diffraction showed no CeOF2 or CeO2.
High-temperature heat-content measurements were made at the Berkeley Thermodynamics Research Laboratory, Berkeley, Calif., on samples of cerous fluoride prepared by the ammonium bifluoride method in platinum dishes and in sintered CeF3 boats at the Reno station.
Fluoride Electrolytes
A prime requisite of a successful molten-salt electrowinning process is a suitable electrolyte. Such factors as cathode, anode, and cell construction materials, composition of the atmosphere surrounding the cell, and voltage and current requirements are also essential but are largely contingent on characteristics of the electrolyte. Accordingly, the electrolyte might be considered the heart of the high-purity-metal electrowinning process.
Surprisingly little basic data have been published on fluoride electrolytes. Because of the key function they perform and the dearth of information on them, considerable effort is being applied to basic studies of electrolytes-the preliminary of cell-box electrowinning investigations.
The electrolyte experimentation is on a much smaller scale than the electrowinning studies; its purpose is to define the physical, chemical, and electrochemical characteristics of combinations of fluoride solvent-phase constituents and of electrolytic baths.
Measurements desired and phenomena about which better understanding are needed concern melting points, viscosity, density, vapor pressure, tendency to wet graphite, solubility of solute in solvent, ionization, transfer of ions, equilibria, polarization, and effects of anode gases.
These investigations on electrolytes require special measuring devices for use with melting and atmosphere-control equipment. Obtaining accurate data is complicated by the extremely corrosive nature of the molten fluorides. Many of these studies are in a formative stage, and a search is still in progress for instruments suitable for making some measurements. However, during the period covered by this report, melting points, conductivities, and solubilities of components were measured, and preliminary electrolyses were conducted on several fluoride baths. The small scale of the operations conserved time and materials in developing suitable electrolytes for trial in the larger cerium electrowinning cells.
Several types of small-scale cells were used for fluoride melting-point determinations and electrolysis of short duration. One cell comprised a carbon pot (welding-rod carbon) 5/8 inch i.d. and 2-¾ inches deep, set inside a 1- by 7.9-inch Vycor glass tube. The anode (a carbon rod 1/8-inch in diameter) and the cathode (a molybdenum strip 1/32 inch wide and 0.01 inch thick) were attached to a 3/16-inch-diameter brass rod and insulated from each other with sheet mica. The brass rod served as the electrical lead to the anode, and a copper wire served as the lead to the cathode. The electrical assembly was introduced into the cell through rubber sleeves set over nipples in a Pyrex glass head. The glass head also had outlets, permitting evacuation and flushing with argon and introduction of a thermocouple protection tube, and was sealed in place in the mouth of the Vycor glass test tube with Apiezon wax. The wax was kept cool with a jet of air.
Neidrach and Dearing have suggested that adding small amounts of chlorides to molten fluoride solvents increases fluidity and electrical conductivity. The described apparatus was used in investigating the effect of adding 5 percent by weight of MgCl2 to part of a CeF3-LiF-BaF2 bath from the Reno cerium electrowinning cell. The MgCl2 lowered the bath melting point from 740° to 690° C., but chlorine gas was produced during electrolysis and anode effect was noted.
The loss of lithium from the CeF3-LiF-BaF2 bath during electrolysis suggested the investigation of a new bath containing less LiF. With the previously described apparatus and the electrode assembly removed, a series of melting points was measured on various CeF-LiF-BaF2 mixtures by the thermal-analysis techniques and visual observations. A bath having a satisfactory melting point of 735° C. was prepared, comprising 77.3 percent CeF3, 12.7 percent BaF2, and 10.0 percent LiF by weight. P. M. J. Gray’s bath contained 26.9 weight-percent LiF.
MgF2 was investigated as a substitute for LiF in the CeF3-LiF-BaF2 bath because it has a lower vapor pressure. The melting points of various CeF3- MgF2-BaF2 mixtures were measured. As the lowest melting point was 930° C., no mixture investigated was suitable as a solvent-phase electrolyte for electrowinning of cerium. However, MgF2 may serve as a bath constituent for electrowinning of uranium ingot and the rare-earth metals melting above 1,000° C.
Using a KF-LiF-NaF solvent-phase electrolyte and adding CeF3, the authors carried out electrolysis in a tantalum cell within a controlled-atmosphere glove box. A pool of molten alkali was noticed around the cathode at the top of the bath during electrolysis. X-ray diffraction patterns of cell products showed that no cerium metal had been produced. With a BaCl2-CaCl2-CeF3 bath, electrolysis in a Vycor cell within the controlled-atmosphere glove box produced massive cerium metal and chlorine gas. Spectrographic analysis showed 0.01 to 0.1 percent calcium in the cerium metal.
Several small-scale cells were designed and investigated to measure the electrical conductivity of molten fluoride systems with alternating current. The fabrication of a suitable cell has been hampered by lack of a nonconducting material resistant to fluoride corrosion.
A cell requiring about 75 grams of bath material was built and gave results in good agreement with values in the literature for the electrical conductivities of molten KCl and NaCl. All parts of the cell in contact with the molten bath were made of graphite or molybdenum, except a Vycor glass tube that provided an insulated electrolyte path between the two molybdenum electrodes, Fluoride corrosion of the Vycor glass prevented good reproducibility in electrical-conductivity measurements of molten fluoride systems with this cell. A new cell is being developed in which the Vycor glass tube is replaced by a hot-pressed boron nitride tube. Data obtained in this laboratory and by other workers have indicated that boron nitride is resistant enough to corrosion by molten fluorides for short periods to serve these experimental purposes.
Vacuum Drying
The constituents for the solvent phase of the cerium electrolyte, that is, CeF3, BaF2 (reagent-grade) , and LiF powder (reagent-grade), were mixed in air in the proper proportions. The mixture was placed in a glass-stopcocked Pyrex flask within a wire-wound resistance furnace. The flask was evacuated to about 10 microns, and the furnace turned on. All vacuum readings were taken with a Pirani gage. As the furnace heated, the flask was pumped out continuously by a mechanical high-vacuum pump with a dry-ice-acetone trap. A furnace temperature of approximately 280° C. and about 15 hours were necessary for the mixture to reach a vacuum of 10 to 15 microns. The powder mixture was then tested for moisture as follows: Moisture removed after dehydration of the electrolyte components was captured in a dry ice-acetone cold trap for 5 to 10 minutes. The trap was then warmed to room temperature and flushed with dry argon through a sensitive moisture-determining instrument, the moisture monitor. When no moisture was detected above the level of the argon blank, the sample was considered dry.
The powder, comprising 73 percent CeF3, 15 percent LiF, and 12 percent BaF2, contained approximately 1 percent moisture before vacuum drying. This mixture will be referred to in this report as the Reno fluoride solvent-phase electrolyte, or the Reno fluoride electrolyte. The procedure removed about 70 percent of the moisture. An experiment indicated that most of the remaining moisture was removed as the temperature was increased to the melting point of the bath. CeO2, the solute phase of this electrolyte, was vacuum- dried in a similar manner.
Electrowinning
Laboratory-Cell Development
Cell Type No. 1
A carbon cell 8 inches high by 4 inches i.d. was designed. P. M. J. Gray used a similar cell. This cell, made of grade CS-31 carbon, was heated externally by a wire-wound resistance furnace through a type-316 stainless-steel can; the inside bottom of the carbon cell was covered with molybdenum sheet that extended 1 inch up the sides. A small 1- by 1-3/16-inch-diameter molybdenum crucible for holding the molten cerium product was placed under the 0.2-inch-diameter molybdenum cathode. The cathode was protected to 1 inch above the bath by a carbon sheath, which also served as an entrance tube for the argon gas that continuously purged the space above the bath. The small molybdenum crucible was suspended from the carbon sheath with molybdenum ribbons so that it could be raised above the molten bath when electrolysis was complete. No evacuation of contained air or occluded gases in the carbon or charge was possible in a cell of this type. A 0.625-inch-diameter, grade CS-31 carbon anode was used with 1-inch spacing between electrodes. The cell was covered with a water-cooled carbon lid, and the lid was covered with 2 inches of transite insulation.
In a cell of this type eerie oxide cannot be added continuously during electrolysis, thus the operation was of short duration or a batch-type one. It is doubtful whether the data obtained can be compared with continuous-operation data in which the oxide feed is added during electrolysis.
Preparation of Charge
A fluoride electrolyte composed of 60.8 percent by weight of CeF3, 26.9 percent LiF, and 12.3 percent BaF2 was used. This mixture was reported to be capable of dissolving 3 to 5 percent by weight of CeO2 at 850° C. The density of the mixture was approximately 4.0 grams per cubic centimeter. The dry-powder mixture required to form a bath 2 inches deep in the cell was calculated to be of the following weight composition:

The cerous fluoride (99.8 percent pure) used in this experiment was prepared from ceric oxide purchased from a commercial source and mixed with ceric oxide prepared from bastnasite in the Extraction Section of the Reno laboratory. The mixture was fluorinated with ammonium bifluoride, as already described. The lithium fluoride powder was reagent grade; spectrographic analysis showing less than 0.1 percent barium, 0.001 percent magnesium, and other metallic and nonmetallic elements as. traces. The barium fluoride was also reagent grade, spectrographic analysis showing less than 0.01 percent calcium, 0.001 percent magnesium, 0.001 percent silicon, 0.1 percent strontium, and 0.001 percent titanium.
The mixed charge was made into briquets in a laboratory hydraulic press at 20 tons per square inch. This permitted charging the complete 1,760 grams of mixture into the carbon cell where the charge was dried for 16 hours at 140° C. After the briquets were charged and the cell was covered, purified argon was passed through the cell for 30 minutes before initial heating.
Melting of Charge
While the purified argon continued to flow through the cell, the furnace temperature was raised. The time required for melting was 4 hours, and the bath was molten at a furnace temperature of 900° C. Furnace temperatures were taken on the outside bottom of the carbon cell with a base-metal thermocouple. The actual temperatures of the molten bath were taken at intervals with a bare platinum-rhodium thermocouple through the port at the top of the lid. Part of the molten bath adhered to a molybdenum strip. This light-blue vitreous slag analyzed 0.1 percent iron, 0.01 percent magnesium, 0.1 percent molybdenum, and 0.001 percent silicon. The slag adhering to the molybdenum strip indicated a bath depth of 2-¼ inches.
Electrolytic Operations
The bath was electrolyzed at a temperature of 820° to 890° C. for 108 minutes. The amperage ranged from 20 to 15 and the voltage between the electrodes from 6.3 to 5.6.
The cathode and the molybdenum crucible were lifted above the molten bath before allowing it to freeze. Purified argon continued to flow through the cell until the bath had reached room temperature, approximately 13 hours.
The molybdenum crucible, which was easily removed from the supporting basket, was filled to the top with nodules of cerium metal in light-blue electrolyte. The cerium metal when cleaned of electrolyte was found by the X-ray laboratory to contain traces of iron and molybdenum. The cathode density was calculated to be approximately 6 amperes per square centimeter. The cerium metal analyzed 0.1 percent barium, 0.01 to 0.1 percent iron, 0.001 percent magnesium, 0.1 percent molybdenum, 0.001 percent silicon, and traces of other metallic elements.
Five separate electrowinning experiments were made with this cell. The temperature of the electrolyte ranged from 890° to 820° C., the voltage between electrodes from 6.2 to 4.4, the amperage from 19.0 to 15.0, and the electrode immersion depth from ¾ to 5/8 inch; the distance between electrodes was held at 1 inch. The time of electrolysis ranged from 1.6 to 3.5 hours per experiment. The following conclusions were drawn: (1) CO2 gas was identified as a product of the electrolysis, and (2) no fluorine gas could be detected as a result of the electrolytic action.
The metal made in these five experimental runs in the No. 1 type cell contained cerium carbide. Considerable electric current was lost through poor insulation between the electrodes and carbon cell top, and it was difficult to keep the electrodes adjusted to their proper positions. The inability to see the molten bath and make necessary adjustments was a major handicap. Purging the space in the cell above the bath with inert gas at pressure did not remove air and moisture, as was indicated by the yellow (CeO2) surface of the solidified bath. The highest current efficiency attained in any of the five runs was less than 35 percent. The melting temperature of the fluoride electrolyte was approximately 715° C.
Cell Type No. 2
The first alterations made on the No. 1 carbon cell were to line it with sheet molybdenum, remove the carbon sheath surrounding the cathode, and improve the transite bushings on the electrodes in the carbon cell top so as to eliminate the stray current losses. The charge to cell No. 2 was the same as that to cell No. 1, comprising the same mixture of CeF3-LiF-BaF2 and ceric oxide. The molybdenum cup was fastened to the bottom of the molybdenum liner and not attached to the cathode.
The bath was electrolyzed at 778° to 814° C. for 178 minutes with the electrodes immersed three-fourths inch. The amperage ranged from 20 to 29 and the voltage from 7.4 to 8.2.
The electrodes and molybdenum cup were allowed to freeze in the bath. The molybdenum lining was eaten through immediately above the bath, and some of the bath contacted the carbon cell. The molybdenum cup was displaced from under the cathode. A small amount of cerium adhered to the molybdenum cathode, and some nodules were dispersed in the frozen bath.
Cell Type No. 3
Inability to observe the cell operations and control the essential factors in Nos. 1 and 2 cells led to the development of cell type No. 3, which was operated in an argon atmosphere. Cell operations were visible through safety glass and controlled by gloved hands through ports in a steel box.
The cell, furnace, and electrode assembly were arranged within a mild-steel glove box. The electrode assembly consisted of a steel frame with an adjustable electrode holder carrying two electrodes and direct-current conductor arms. The fluoride bath was kept molten by external heating with a resistance furnace made of nichrome wire wound in a 5-inch-i.d. alundum sleeve. The box atmosphere was pumped down and purified argon introduced before melting the “Gray” electrolyte. The argon atmosphere was tested with a gas master instrument and moisture monitor to control purity. One 0.375-inch-diameter carbon-rod anode and one 0.200-inch-diameter molybdenum-rod cathode were used.
Iron, Vycor glass, carbon, and graphite were investigated as cell materials. Cerium metal prepared in the externally heated iron cell, but not in contact with the iron, contained 5.27 percent iron. Vycor glass was corroded with the molten fluoride bath and the cerium metal. Cerium metal that had contacted the carbon or graphite cell walls or bottom contained up to 2.2 percent carbon.
When molten electrolyte and cerium metal contacted the CS-31 carbon cell walls and bottom, cracks developed, resulting in loss of bath. AGX-grade graphite was substituted for CS-31 carbon and did not crack under similar conditions.
Cell Type No. 4
Preventing the molten cerium from contacting the graphite cell walls and bottom was investigated, using the furnace and electrode assembly described in the cell type No. 3.
In one experiment with the Reno fluoride solvent-phase electrolyte, a sintered cerous fluoride disk was placed in the cell bottom. The disk prevented the cerium metal from contacting the graphite cell, and no evidence of carbide formation was noted. However, the CeF3 disk dissolved slowly in the bath.
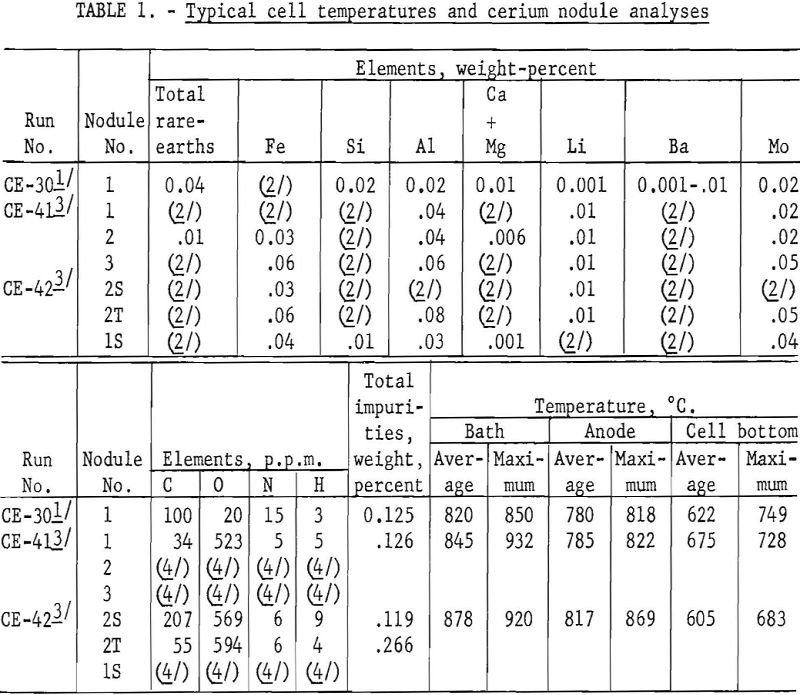
In another experiment (CE-30), an air-cooled copper coil was placed under the graphite cell bottom and a frozen layer of electrolyte maintained. Two hundred grams of massive cerium metal was deposited and held above the cell bottom. Analysis of a nodule of CE-30 cerium metal by the Boulder City station showed 0.01 percent carbon. Table 1 gives analyses of this nodule for metallic and nonmetallic impurities.
A mixture comprising 1,178 grams of the Reno fluoride electrolyte and 1,622 grams of old bath was dried using the procedure described under vacuum drying, page 9.
This bath was melted in a 4-inch-i.d. by 4-5/8-inch-deep graphite cell, using a wire-wound resistance furnace within the controlled atmosphere and temperature cell box. A bath temperature of 112° to 850° C. and a cell-bottom temperature of 590° to 749° C. were maintained during electrolysis.
A 0.200-inch-diameter molybdenum-rod cathode and a 0.625-inch-diameter carbon anode, three-fourths inch apart, were immersed 1 inch in the molten bath.
Ceric oxide was added to the molten bath at a rate of approximately 2.5 grams per 5 minutes during the 510 minutes of electrolysis, and an average current of 19.6 amperes and 6.0 volts was maintained.
Reno Cerium Electrowinning Cell (Cell Type No. 5)
As a result of previous data semicontinuous cell type No. 5 was developed. It differs mainly from the earlier types in that the electrode arrangement for melting the bath is the same as that for electrolysis. Although this 6-inch-diameter graphite cell is the latest cell development reported at this time, plans have been made to develop a continuous 12-inch-diameter electrowinning cerium cell in the near future.
In order to have more positive control of the temperatures of the cathodes, anodes, cell walls, and cell bottom and maintain a layer of frozen electrolyte on the cell walls and bottom, a triagonal electrode arrangement adapted to internal melting was designed and built. A 40-volt, 200-ampere, silicon, a.c.-d.c. rectifier unit and a 300-ampere, 40-volt, a.c. arc-welding unit were used as power sources. The No. 5 cell was operated in a controlled atmosphere and temperature glove box. Figure 1 shows the principal features of this cell and glove box.
The meltdown of the Reno fluoride electrolyte was initiated by passing current through the electrodes and through graphite-ring resistors that contact the bottom ends of the electrodes. The resistors are set on minus-10-mesh old-bath material within the graphite cell and are removed after enough bath has melted to carry the current.
Electrowinning Cell and Electrode Assembly
The electrowinning cell is AGX-grade graphite, 6-1/8 inches inside top diameter by 4-½ inches deep. The cell tapers to 6 inches i.d. at a depth of 2-¼ inches and to 3-½ inches i.d. in the remaining 2-¼ inches. The graphite cell walls are ½ inch thick and the bottom 1 inch thick. One thermocouple well is drilled longitudinally in the graphite cell wall; another thermocouple well is drilled in the graphite cell bottom. Chromel-alumel thermocouples in sillimanite protection tubes are placed in the thermocouple wells.
The cell is insulated on the sides with 4 inches of insulating brick with ½ inch of minus-4-mesh fire-clay brick grog between the graphite and the insulating brick. The bottom of the cell rests on 1-½ inches of insulating brick, allowing more heat loss and helping to maintain a bottom layer of frozen bath or skull. The cell cover is fire-clay brick 2-½ inches thick. The central brick of the cell cover is drilled to accommodate electrodes and feed tube. A 3/8-inch-i.d. sillimanite tube with a Pyrex funnel for feeding cell charge materials and ceric oxide terminates at the bottom of this brick cover.
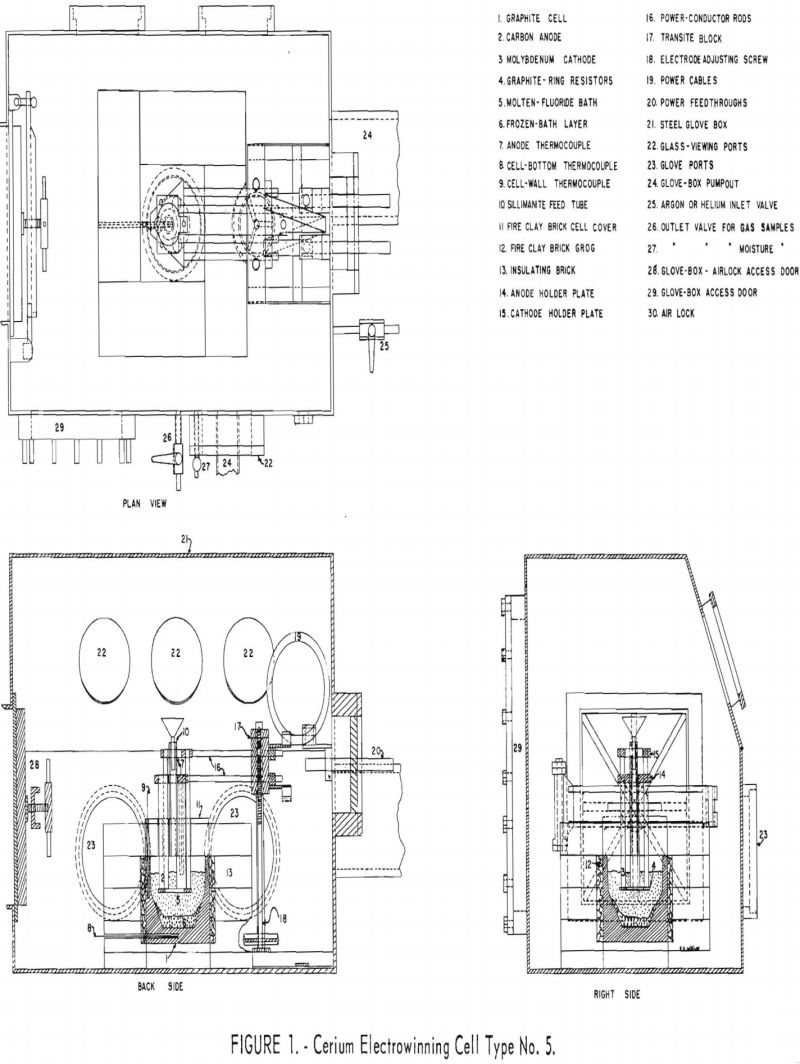
The electrode assembly consists of an adjustable three anode-three cathode triagonal system. (See fig. 1.) A thermocouple well is drilled longitudinally in a carbon anode within 1 inch of the bottom. A chromel-alumel thermocouple in a sillimanite protection tube is placed in the anode thermocouple well.
Two copper power-conductor rods from the anode and cathode holder pass through a transite block that is raised and lowered on a steel frame by operating an adjusting screw to position the electrodes vertically.
Rubber-insulated power cables carry the current to the conductors from power feedthroughs, which are vacuum-sealed in a rubber-gasketed Micarta plate on the end of the glove box. Rubber-insulated power cables carry current from the a.c.-d.c. rectifier and a.c. welder heater to the ends of the power feedthroughs outside the steel glove box.
Steel Glove Box
The electrode assembly and graphite cell are centrally located in the controlled atmosphere-temperature-pressure (C.A.T.P.) steel glove box. The glove box and air-lock enclosures are made of welded ¼-inch mild-steel plate. (See figs. 1 and 2.) The inside dimensions of the glove box are 24 inches wide by 36 inches long by 26 inches high. The air-lock inside diamensions are 12 inches wide by 12 inches high by 18 inches long. The room air-lock door and the glove box-air lock access door are sealed with flat rubber gaskets. A 10- by 18-inch mild steel door is sealed to the rear side of the glove box with a flat rubber gasket. This glove-box access door is removed for setting up and dismantling the electrowinning cell and electrode assembly.
The inside of the glove box and air lock were sandblasted and painted with a water solution of 1-percent “Siliclad.” No noticeable corrosion had taken place on the inside surfaces of the mild steel glove box after exposure to vapors from various melting and electrolytic experiments with fluoride baths over a period of 9 months.
The 1-inch-thick, laminated, safety-glass viewing ports were slightly etched after several electrolytic runs and were removed and buffed to restore transparency. A 100-watt light bulb within the box was used for illumination.
Replacing Air with Inert Atmosphere
Figure 2 shows the oil-diffusion pump, a welded-steel cold trap, and a mechanical fore pump connected to the end of the glove box opposite the air lock. The 6-inch-diameter oil-diffusion pump and the mechanical fore pump can be used to evacuate the glove box through a 6-inch steel pipe and the air lock through a 4-inch steel pipe. The box and air lock can also be evacuated through a 2-inch pipe attached directly to the mechanical pump.
Argon or helium is admitted to the glove box through the ¼-inch copper tube shown in figure 1. Box atmosphere pressures were measured through this tube using a mercury manometer. Samples of box and air-lock atmospheres for gas master tests and Orsat gas analyses were obtained through the ¼-inch valve shown in figure 1. Box atmosphere samples for moisture determinations were transferred into the moisture monitor through a copper Teflon tube system connected to a vacuum pump.

Melting of Bath
Electrocerium fluoride charges were melted by passing direct current through the graphite resistors contacting the bottoms of the electrodes. The 40-volt 300-ampere, silicon, a.c.-d.c. rectifier unit was the d.-c. source. Arcing was noted with erratic voltage and current conditions. Moreover, cerium carbides were formed, as cerium-metal deposition began with the initial melting of the charge when graphite resistors were present. Analysis of a cerium metal sample, where part of a graphite resistor remained in the bath during electrolysis, showed 5,200 p.p.m. carbon.
Several attempts were made to use cerium metal strips as resistors in melting with direct current. In all instances the cerium metal melted, breaking the circuit before enough molten bath had been obtained to carry the current. Additional work on this problem is planned in the future.
Cerium Electrowinning Run (CE-42)
A cerium electrowinning run was made using the Reno fluoride solvent phase of the electrolyte and CeO2 solute with a.-c. internal meltdown in the type No. 5 cell.
The C. A. T. P. glove box containing the triagonal electrode assembly and the 6-inch-diameter insulated graphite cell described previously were pumped down to 75 microns. High-purity tank argon was run into the glove box to ¾-p.s.i. The box atmosphere and high-purity tank argon were compared with the gas master and found to have equal thermal conductivities.
Three thousand one hundred and thirty grams of minus-10-mesh charge, made from the old bath of a previous electrowinning run and vacuum dried, were loaded into the cell. Two graphite half-ring resistors, 3/16 inch thick by 3-¾ inches o.d. by 3/8 inch wide, were placed on top of the minus-10-mesh charge. The electrodes were lowered to contact the resistors. The resistors were placed 1-¾ inches from the top of the cell, and 1 inch of vacuum-dried, Reno fluoride, solvent-phase electrolyte powder was placed on top of the resistors. When the graphite resistors were placed on fresh powder, arcing was caused by shrinkage of the bath.
Alternating current at an average of 7 volts and 300 amperes was passed through the graphite resistors for 2 hours using the 40-volt 300-ampere arc-welding unit. About -1,500 grams of Reno fluoride solvent-phase electrolyte was added to the cell by the gloved operator through the feed tube during the alternating-current meltdown. No arcing was noted. When a fluoride bath was obtained around the electrodes and the bath temperature was 750° C., the graphite resistors were removed, using molybdenum tongs.
The current was then switched from a.c. on the welding unit to d.c. on the 40-volt, 200-ampere silicon rectifier. Ceric oxide (CeO2) powder was charged into the molten bath by the gloved operator through the sillimanite feed tube, using a calibrated glass spoon. A ceric oxide feed rate of 2 grams per minute was maintained during the 146 minutes of electrolysis.
An average direct-current voltage of 6.7 and an average amperage of 203 were maintained during electrolysis. The electrodes were immersed in the bath three-quarters inch, giving an initial cathode current density of 11.3 a. per sq. cm. and an initial anode current density of 4.5 a. per sq. cm. Adjoining cathodes and anodes were 0.75 inch apart and 1.25 inches from the cell wall.
During electrolysis the temperature of the anode having the thermocouple well averaged 817° C. and the cell-bottom temperature 605° C.
Samples of the cell-box atmosphere were taken for Orsat gas analysis at approximately 15-minute intervals. At intermittent intervals the box was partly pumped down, and high-purity argon was introduced to maintain the box atmosphere at about 66° C. which enabled the operator to work in the box. A maximum of 17.6 percent CO2 and 1.8 percent CO was reached in the box atmosphere. Usually, the pumpdowns and introduction of argon maintained the box atmosphere below 12 percent CO2 and 1 percent CO. Qualitative tests of box-atmosphere samples for fluoride ion, using zirconium alizarin solution, were negative.
Some of the bath foamed over the side of the graphite cell during this run. A small amount of a white, powdery sublimate collected on the anodes above the bathe Previous X-ray diffraction patterns indicated that this sublimate was chiefly LiF. The glass viewing ports were also coated with a white, powdery sublimate and were slightly etched.
The bath with immersed electrodes was allowed to freeze in the cell-box atmosphere upon completion of the run and remained there for 15 hours. The frozen bath was then readily lifted from the graphite cell by means of the electrodes. The graphite cell was not visibly corroded.
Six hundred and six grams of massive cerium metal nodules, mostly ½- by ½- by 1-inch, were found scattered in the frozen bath beneath the electrodes. None of the cerium nodules touched the electrodes, walls, or bottom of the graphite cell. Only a very small amount of cerium metal adhered to the molybdenum cathodes.
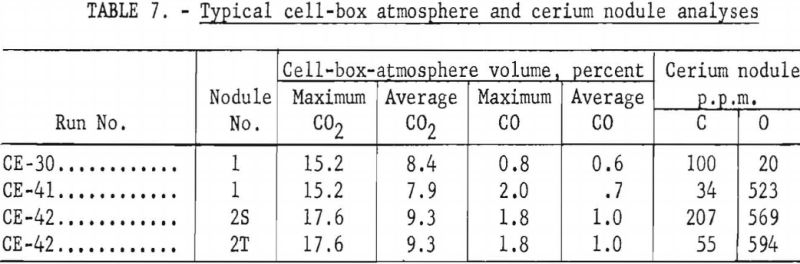
Each cerium metal nodule under molybdenum cathode No. 2 in electrolytic run CE-42 was filed in the air to obtain one or two bright surfaces. The filed surfaces were examined megascopically after the nodules had remained in air 40 hours. Some nodules were tarnished and others remained silvery. Table 2 compares a silvery-surfaced nodule, 2S, with a tarnished-surfaced nodule, 2T.
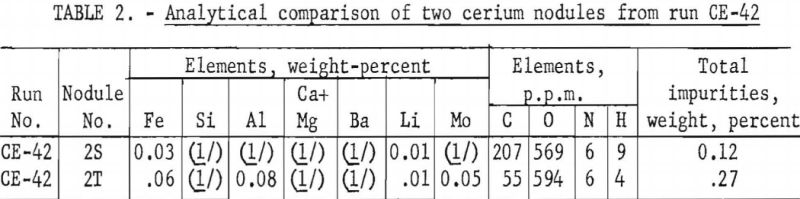
This method has been used to classify low- and high-grade cerium metal nodules from several electrolytic runs. Generally, nodules with silvery surfaces are higher grade than those with tarnished surfaces. Investigation of the air-corrosion classification method is being continued.
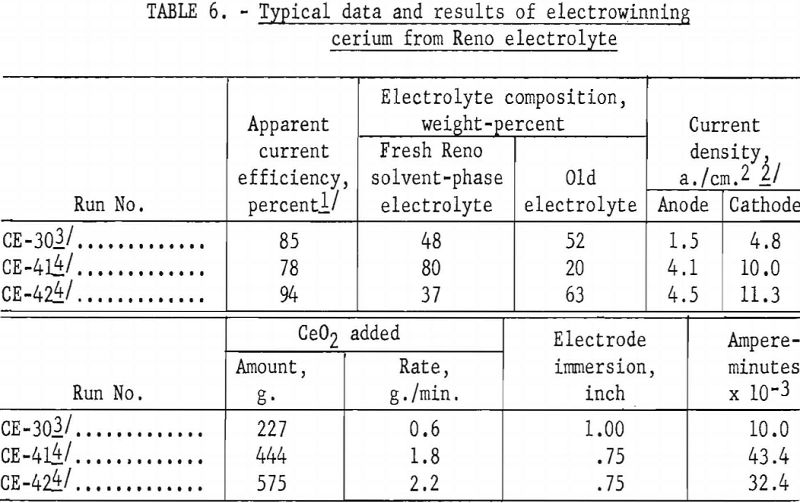
Current efficiency, computed from massive cerium metal produced, was 94 percent. Current efficiencies ranged from 70 to 94 percent, depending upon the amount of old bath reused in the charge and other factors inherent to small research cells and short electrolyzing periods.
Analyses of Cell-Box Gases
Gas analyses were performed during each run in cell type Nos. 3, 4, and 5. Gas samples were removed from the cell box by means of an evacuated gas-sample bottle 10 inches long and 1-½ inches in diameter with a stopcock at both ends. The sample bottle was evacuated to 20 microns pressure or less, then attached with a short piece of rubber tubing to a valve near the top center of the glove box. After the tubing was flushed with argon to remove any air, the valve and stopcock were opened and a sample of the glove-box atmosphere was sucked into the sample tube. The sample was then transferred to an Orsat apparatus and analyzed for CO2, CO, and O2 by standard Orsat procedure. The Orsat apparatus had a capacity of 50 ml., and gas volumes could be measured to 0.1 ml. or 0.2 percent of the total gas volume. Qualitative tests were also made for HF and F2 by bubbling samples of the box gas through a zirconium-alizarin solution prepared according to Feigl.
The major gaseous product of the electrolysis was CO2, and small amounts of CO were present. In this respect the current cerium cell is similar to the aluminum cell, in which CO2 is known to be the principal anode gas and CO is formed by the reduction of CO2 by anode carbon or by metal fog. The amount of CO seldom exceeded 2 percent of the total volume of the gas in the glove box and usually remained within a range of 0.8 to 1.6 percent, despite the length of the runs. The concentration of CO2 continued to build up as the runs proceeded. By partly evacuating the glove box and adding pure argon at intervals, the operator diluted the CO2 and CO and controlled the box atmosphere. The maximum concentration of CO2 permitted in any run was 18 percent. The correlation between CO2 content of the box atmosphere and metal purity is to be investigated. Possibly, a greater CO2 concentration may be tolerated.
CF4. has been reported in the off gases of both aluminum and uranium cells and would be expected to be present in preference to F2 in any electrolysis of a fluoride bath using a carbon anode at temperatures greater than 450° C.
In the uranium cell the CF4 was usually present in concentrations of 0.19 percent or less, and in the aluminum cell the gas was detected only during an anode effect caused by depletion of Al2O3 in the cell.
There are no convenient chemical tests for the fluorocarbons, because of their extreme inertness. It is planned to have mass spectrometric analyses made of the cerium cell gas. Attempts to trap out CF4 by means of liquid oxygen traps did not indicate the presence of the gas in detectable quantities. It is believed that some CF4 and possibly other higher fluorocarbons may be present in the off gases of the cell but in too small amounts to be detected by available procedures.
Oxygen has been detected occasionally in the box gas sample, but its presence has usually been traced to small leaks in the glove box that developed after the run had begun.
At least 1 percent HF has been reported in the off gases of the aluminum cell, due to moisture in the fluoride baths and hydrogen from anode hydrocarbons. No HF or F2 has been detected in the off gases from the Reno cerium cell. However, the slow etching of gas-sample tubes, glass viewports, and other pieces of glass apparatus seems to indicate the presence of at least small amounts of HF.
Typical moisture analyses of the high-purity argon and helium gave moisture contents of less than 10 p.p.m. by volume.
The purity of the cell-box gas before melting and electrolysis was determined by an instrument that compares the thermal conductivity of a sample gas with that of a standard gas. High-purity argon and helium were used as standard gases. When the thermal conductivities of the box gas and tank gas matched, the box atmosphere was considered to be of proper purity.
Lithiothermic Reduction and Vacuum Refining
Anhydrous cerous fluoride and chloride have been used by previous investigators in preparing cerium regulus by metallothermic reductions. P. M. J. Gray obtained only finely dispersed cerium regulus in the lithium-bomb reductions of cerous chloride in molybdenum cans. In similar reductions of cerous fluoride ce obtained buttons of massive cerium regulus. He attributes the failure of the dispersed metal to coalesce in the chloride reduction to an oxide or oxychloride skin on the metallic surface. This skin is derived from small amounts of water retained by the cerous chloride after dehydration or picked up from the air during loading.
In the Reno experiments cerous fluoride was chosen owing to the relative ease of preparing it in an anhydrous form and its stability in air at ordinary temperatures. Cerous fluoride also has a lower vapor pressure than cerous chloride. Lithium metal was chosen as the reductant because the LiF slag has a low melting point (870° C.), thus facilitating coalescence of the cerium.
Calcium and magnesium fluorides melt at 1,360° and 1,396° C., respectively. Cerium from the calcium reduction of cerous chloride contains up to 2 percent calcium and magnesium forms alloys with cerium.
Massive cerium regulus was prepared by the lithium-iodine reduction of anhydrous cerous fluoride in welded molybdenum and tantalum cans, lime-lined steel-pipe reactors or bombs, in a Globar resistance furnace. P. M. G. Gray reported that cerium metal prepared by lithium-bomb reduction in a molybdenum can had negligible molybdenum pickup. Spedding found that cerium prepared by calcium reduction in tantalum crucibles contained less than 0.05 percent tantalum. As much as 2.7 percent molybdenum and 0.3 percent tantalum were reported by X-ray fluorescent analysis in Reno bomb cerium regulus prepared in molybdenum and tantalum cans.
Because the vapor pressure that builds up in a properly fabricated molybdenum or tantalum container is a primary factor in bomb reduction efficiencies, techniques for fabricating and welding molybdenum and tantalum cans for reduction containers were developed in these laboratories. The cans were welded in a dry argon atmosphere in a glove box using a special direct-current tungsten arc welder.
A procedure was developed for preparing 50-gram buttons of cerium regulus by bomb reduction. The cerous fluoride made by the thermal fluorination of ceric oxide with ammonium bifluoride was heated under a vacuum of 42 microns 300° to 400° C. for 12 hours to remove moisture and other gases. A tantalum or molybdenum can, 1-¾ inches in diameter by 2-½ inches in height, was made from 0.005-inch sheet with a welded bottom and side seam. The can was degassed in the induction furnace, then transferred to an argon atmosphere glove box and loaded with thin alternate layers of charge comprising 78.17 grams of CeF3, 9.6 grams of silvery Li metal, and 20 grams of resublimed I2. The purity of the glove-box atmosphere was controlled by comparing the atmosphere with high-purity tank argon using the gas master.
A molybdenum or tantalum plug was placed in the top of the loaded can, and the can was packed in a wrought-iron pipe reactor 2-¼ inches i.d. by 8-¾ inches high, with a liner of recalcined lime. The pipe reactor had a welded bottom and was sealed with a threaded pipe cap using litharge and glycerine.
The bomb was removed from the dry-argon-atmosphere glove box and charged in the Globar furnace at 1,280° C. If reduction was begun with a cold furnace, a finely dispersed cerium metal in a lithium fluoride-lithium iodide slag was produced. The outside of the pipe was maintained at 1,100° to 1,150° Co for 40 minutes, then the reactor was removed from the furnace and air-quenched for 15 hours before opening.
Recoveries of massive cerium regulus as a button ranged from less than 50 to 95 percent. The lithium-fluoride-lithium-iodide slag broke away readily from the cerium button. When a molybdenum can was used, the molybdenum adhered to the cerium, making it necessary to machine the molybdenum from the cerium button. When a tantalum can was used, the tantalum could be peeled away readily from the cerium button, however, the can was sacrificed in the reduction.
Leaks in the plugs of the molybdenum or tantalum can and voids in the lime packing, which allow the reaction gases to force unreduced charge out of the reduction zone, are believed to be the principal causes for the failure to reproduce consistently high reduction efficiencies. Extreme care is required in making and loading molybdenum or tantalum containers and bombs.
Cerium reguli from the lithium-iodine bomb reductions of cerous fluoride in tantalum and molybdenum cans were melted and refined using quartz-tube vacuum induction and vacuum resistance furnaces.
The tantalum and molybdenum cans, 1 inch in diameter by 4 inches high, were fabricated in the Reno laboratories from 0.005-inch gage sheet and welded in an argon-atmosphere glove box. They were degassed in the quartz-tube induction furnace.
Several pieces of bomb cerium regulus were scraped clean of slag and oxide with a molybdenum knife and loaded into a degassed tantalum or molybdenum can in an argon-atmosphere glove box. Then the loaded can was transferred under argon to the 3-inch-diameter quartz-tube induction furnace.
To facilitate better low-range temperature control in the vacuum induction furnace, a secondary coil was connected in series with the furnace coil. This coil allowed degassing of the cerium regulus below its melting point and prevented ejection of molten cerium from the can by the sudden evolution of gases. Vacuum refining experiments also were performed using a resistance furnace surrounding the quartz tube to bring the cerium regulus slowly from room temperature to its melting point.
In a typical vacuum refining experiment, the cold quartz tube was evacuated to 3 microns and the temperature of the cerium regulus, measured by optical pyrometer, was varied from 740° to 920° C. in a degassing period of 4.65 hours. The vacuum rose from 3 to 230 microns and dropped to 15 microns at the end of the experiment. When Reno electrocerium metal was vacuum-refined, as previously described, the vacuum varied by only a few microns.
Several pieces of cerium regulus had melted into one ingot with several blowholes at the top. Machining was necessary to remove the molybdenum, but the tantalum could be peeled readily from the refined cerium ingot. Some of these round ingots were cold-rolled in an argon atmosphere and shaped into 0.1- by 0.2- by 1-inch specimens for electrical conductivity investigations. In table 3 a Reno bomb cerium regulus, before and after vacuum refining in a tantalum can, is compared analytically with Reno electrocerium and a commercial cerium metal.
Ceramics
Titanium oxynitride crucibles were prepared and used at Reno for vacuum refining commercial cerium metal. L. S. Foster describes titanium and zirconium oxynitride crucible preparation and cerium melts under vacuum in these crucibles. According to Foster, cerium does not attack the crucibles, but no data are given on contamination of the cerium.
A mixture of 70 percent titanium nitride and 30 percent titanium oxide by weight with Carbowax was molded under pressure in a steel die. The green crucibles were calcined at 250° C. to remove the Carbowax binder, then fired in a dry argon atmosphere in the induction furnace at 1,550° to 1,600° C.
Commercial cerium metal was vacuum-melted in the titanium oxynitride crucibles in the quartz tube using an induction furnace. Approximately 20 grams of cerium was melted in each crucible, and temperatures of approximately 1,000° C. were attained. The cerium adhered to the crucible walls and bottom, and spectrographic analysis indicated contamination of the cerium with titanium at an approximate weight-percent of 0.1 to 0.01 titanium.
In summarizing his experimental results, Foster states that CeS is in no way superior to TiN or ZrN for melting cerium.
Some grades of graphite have proved satisfactory as cell construction materials at 810° C. in the Reno cerium electrowinning runs. Unlike Vycor glass and iron they were not attacked by the molten fluoride electrolyte. As the graphite surface was not wet with the Reno fluoride electrolyte, the frozen electrolytes could be easily removed, making possible reuse of the graphite cells.
In cerium electrowinning runs using cells of a certain grade of carbon, carbides were formed and the cells cracked when molten cerium contacted the carbon, resulting in loss of the molten electrolyte. Under parallel conditions in graphite cells, the molten cerium formed carbides on contacting the graphite, but no cracking took place at temperatures of 810° C.
When a frozen layer of electrolyte is maintained next to the graphite, as in cell type No. 5, electrocerium low in carbon impurity can be prepared in a graphite cell under properly controlled atmosphere and temperature conditions.
The development of laboratory use of CeF3 as a ceramic liner in the fluorination tube furnace at Reno is reported on page 6.
Electrical Conductivity of Cerium Metals
The Bureau’s Physics of Metals unit has expended much of its effort on the design, fabrication, and operation of equipment to give reproducible values of electrical conductivity of cerium from room temperature to that of liquid oxygen. The purpose is: (1) To assess the relative purity of samples of electrocerium and (2) to determine the true electrical conductivity of cerium over this range of temperature. Progress and development toward these goals are as follows:
The relative purity of the cerium samples can be measured by their conductivity, as the electrical resistance of most elements of atomic numbers 57 to 71 decreases with temperature, although not linearly, finally reaching a value independent of temperature. This low-temperature residual resistivity is a function of the purity of the metal and the strains and dislocations in the sample. Thus, if the strains and dislocations can be minimized or held approximately constant, electrical conductivity will be a very sensitive measure of the purity. Control of strain level and dislocations is not too formidable a problem, as purity is by far the most sensitive parameter according to previous investigators.
In addition to the complications imposed by impurities, the electrical conductivity of cerium is further complicated by at least three reversible allotropic changes between its melting point and absolute zero. A study of the literature shows that the inner state of the metal is not one of equilibrium but of simultaneous coexistence of two or more of these changes.
The equipment is being developed so that a wide range of temperature rates will be possible; thus, the area of the hysteresis loop can be investigated as a function of the temperature rates.
The Reno electrocerium is usually spectrographically free of calcium and magnesium, two major impurities in the cerium used by most investigators. As previous studies of the allotropic transformations have revealed that calcium and magnesium are particularly interfering impurities, the values of electrical conductivity obtained in this investigation may be of considerable significance.
Correlation of this type has been done on other elements with great success. For instance, Bell Telephone Laboratories reports in a recent article on low-temperature resistance of “varistor”-type high-purity copper:
It is planned to use the foregoing correlation as an aid in selecting some varistor coppers in the near future. However, while this method may prove useful in selecting varistor coppers, the present results are reported primarily to call attention to a particular example of the practical utility of low-temperature resistance measurements in assessing the relative purity of metals, since it is felt that the method may have more general utility as a criterion of purity than is generally recognized.
The importance of obtaining the true electrical conductivity of cerium, is evident from examining the atomic structure of the rare-earth elements. They have similar electronic structures, although their nuclear structures differ. As the atomic weight of the series increases there is, with few exceptions, a regular decrease in atomic volume.
The chemical reactivities of the rare-earth elements, which are a function of their electronic structures, are quite similar. Their physical properties, which depend on their nuclear structures as well as other factors, show significant differences. Thus, they offer an excellent opportunity for checking present theories of metals and possible correlation of the properties of the metals and their structures. However, as they are extremely reactive and small amounts of impurities can drastically change their properties, accurate reproducible measurements of their characteristics are generally lacking.
Investigations With Chamber No. 1
The first chamber was fabricated from 3-inch-i.d. by 7-inch-long steel pipe, welded shut at the bottom, and a 3-inch-i.d., 5-inch-o.d. O-ring flange welded to the top. The cover was a 5-inch-diameter Bakelite disk ½ inch thick, to which the entire bridge circuit was rigidly attached. The bridge comprised separate current and potential contacts with a copper-constantan thermocouple at either end of the sample. The sample was held tightly against the contacting edges by a spring-loaded Bakelite sheet. The cover also contained an outlet for evacuating the test chamber and a ½-inch-diameter brass rod for supporting the chamber in the Dewar of liquid oxygen. Vacuum-tight electrical connections were made by sealing the leads in a cold-setting plastic where they pass through the Bakelite cover.
Temperature of the sample was controlled by variable electric heaters, one in a thin-walled copper tube connecting the sample block to the bottom of the test chamber and another between the O-ring flange and the Bakelite cover. A Kelvin bridge circuit was chosen to measure the resistivity, because the null detector method of measuring would eliminate errors due to contact resistances. These resistances will be present when working with reactive metals such as cerium, and because the total resistance of the sample is small the errors would be large.
The bridge consists of separate current and potential contacts to the sample. The potential drop across the sample is measured by a Rubicon type-B precision potentiometer; the current, which is supplied by five 6-volt, low-discharge-type storage batteries, is measured by the potential drop across a standard resistance of 1 ohm.
The resistance, R, of the sample is given by Ohm’s law as the quotient of the potential drop and current. The resistivity, ρ, of specimens is then computed from the relation ρ = RA/L where A is the cross-sectional area and L the distance between potential contacts. The temperature of the sample is indicated by two copper-constantan thermocouples, one at either end of the sample.
Specimens for comparing electrical conductivity were made originally as follows: Samples of Reno electrolytic and bomb cerium metals and commercial cerium metals were vacuum-melted in degassed, ½-inch-i.d. tantalum cans. Then these samples were prepared in an argon atmosphere in the glove box for X-ray lattice constant and electrical conductivity measurements. Disks ¼ inch thick were cut with bolt cutters from the ½-inch-diameter cerium cylinders. The disks were cold-rolled into strips 1/8 inch thick with a hand rolling mill. The strips were squared into approximately 0.2 by 0.1 by 1-inch samples with a steel file and polished on grit 1/0 emery paper. All machining, rolling, polishing, and sample preparation were performed in an argon atmosphere.
Conductivity values obtained in chamber No. 1 on samples of cerium produced by three methods showed a distinct correlation with purity. (See table 4.)

The apparatus, although useful in gathering preliminary data, had several disadvantages: (1) It was extremely difficult to achieve zero flow of heat at the start of the run without considerable fluctuation in temperature. As the resistivity of cerium is a function of the thermal history of the metal, it would be necessary to duplicate this cycle every run. (2) The temperature gradient across the sample could not be held at the desired maximum of ½° C., because the heat was being exchanged primarily from either end of the sample. (3) The danger of thermoelectric effects was present, as the bridge circuit was not in an isothermal region. (4) Finally, it was very difficult to reproduce the thermal cycles.
After some effort was made to eliminate these faults from the equipment, it became apparent that a drastic change was needed in the basic design, and chamber No. 2, was designed and constructed. This chamber proved very successful.
Investigations With Chamber No. 2
Chamber No. 2 was fabricated from high-conductivity copper and has provisions for running three samples simultaneously. The chamber is surrounded by several inches of polystyrene foam, and temperature is controlled by circulating gases at various temperatures through coils soldered to the outside of the container. The same bridge circuit used with chamber No. 1 was also used with chamber No. 2. A technical paper, describing the equipment in detail, is being prepared for publication. The use of a small jeweler’s lathe in making the samples did not introduce as many dislocations as rolling.
As it is impossible at present to remelt cerium without introducing impurities, the length of the conductivity samples is limited to that of the nodules. The average conductivity sample is approximately 0.125 inch in diameter and 1 inch long; thus, uniformity of cross section and measurement of the 0.8 inch between potential edges represent the largest uncertainties in the computed conductivity, being plus or minus 1.5 percent.
An axis is chosen through the longest dimension of the irregular nodule so that the finished sample will be as long as possible. A cold-setting plastic is cast around the nodule in a ¾-inch cylindrical tube with the chosen axis of the nodule coinciding with the axis of the cylinder. The cylinder is mounted in a jeweler’s lathe and machined under vacuum oil to approximately 0.135 inch in the section containing the metal, leaving plastic knobs on the ends for mounting for the final cut. The semifinished samples are then stored in a desiccator. Just before measurement they are mounted again in the lathe and machined to 0.125 inch plus or minus 0.0005 inch, then mounted in the inert-atmosphere test chamber. Although much work remains to be done on controlling grain size, orientation, stress level, and dislocations the polycrystalline samples, preliminary investigations indicate that these factors were approximately constant in the samples used in this investigation.
By means of chamber No. 2 it was possible to compare Reno electrocerium with commercial cerium at liquid-oxygen temperatures. Samples for the comparison were machined, then measured simultaneously. A constant temperature rate of 0.2° C. per minute was used throughout the experiment, and the samples were held at the low point for 36 hours to insure equilibrium before the return cycle was begun. As was true with chamber No. 1, the conductivities with chamber No. 2 were greater for the purer metal. (See table 5.)

No direct comparison can be made between the data from experiments in chamber Nos. 1 and 2 because of differences in temperature and sample fabrication. The large temperature gradient in the bridge circuit of chamber No. 1 probably affected the values obtained.
No low-temperature transition was evidenced in the commercial metal, nor was there any hysteresis. Reno electrolytic cerium (CE-42-2S) showed a normal transition and hysteresis pattern with the curves rejoining at minus 90° C. The measurement given for CE-42-2S is valid only for this sample, as the nodules produced in the same run vary. For example, another sample, CE-42-2, had a conductivity of 18.0 x 10², ohm-¹ cm.-¹.
Discussion
High-purity cerium ingot was prepared in Nos. 4 and 5 cells by maintaining a cell liner of frozen electrolyte and a controlled argon-carbon dioxide-carbon monoxide cell atmosphere. Control of the anode and cell-bottom temperatures in the cerium electrowinning runs given in tables 1 and 6 prevented molten cerium from contacting the graphite.
Analyses for metallic, nonmetallic, and gaseous impurities show that the cerium nodules from the same electrowinning run, as well as those from different runs, vary widely in composition. (See table 1.) Each set of analyses represents only the nodule sampled and illustrates the extreme reactivity of cerium metal.
The authors believe that the carbon in the cerium comes from the CO2 released at the anode. The variation in carbon content of the nodules shows the need for careful control of the composition of the cell-box atmosphere and the temperature and viscosity of the electrolyte. For example, samples of cerium metal nodules from CE-30 were analyzed for carbon at the Rolla (Mo.) station of the Federal Bureau of Mines, using the conductometric carbon analyzer. Ten determinations averaged 1,590 p.p.m. carbon with a low of 20 p.p.m. and a high of 6,200 p.p.m. The nodules were exposed to the atmosphere, and pieces picked from nodules having the least surface corrosion on four determinations averaged 35 p.p.m. carbon with a low of 20 p.p.m. and a high of 50 p.p.m.
The aluminum, silicon, and iron impurities in electro-cerium are attributed mainly to the fire-clay-brick cell cover and the carbon-welding-rod anodes. Iron also may be introduced in crushing old electrolytes for reuse. Lithium and barium impurities in cerium are low, although their fluorides are solvent-phase electrolyte constituents. Molybdenum cathodes and a molybdenum tool used to remove the graphite resistors after meltdown are the sources of molybdenum contamination of the electrocerium.
During the 3.65 hours of electrolysis it is doubtful whether the electrolyte reached a state of equilibrium, regarding either oxyfluoride content or cation versus anion balance. The considerable difference in the analysis of separate cerium nodules indicates that the selective purification of the electrolyte did not proceed to completion.
Before bath temperatures, cell-box-atmosphere temperatures, pressures, bath equilibrium, and compositions can be correlated with the amounts of impurities in the electrocerium, electrolytic runs of 72 hours or more are believed necessary. A cerium electrowinning cell has been designed for semi-continuous operation of 72 hours or more. A mechanical feeder for CeO2 will be used in operating this cell.
Coalescence depends upon the surface condition of the metal and upon surface tension, viscosity, and relative density of metal and electrolyte, manner of feeding ceric oxide, and possibly other factors. The authors believe that longer runs in a larger cell, with a deeper zone of electrolyte at a temperature above the cerium melting point, would aid in nodule coalescence.
The work in the Reno laboratories has been confined solely to fluoride electrolytes, the electrowinning of cerium from CeO2, and the lithiothermic reduction of CeF3. The problem of rare-earth metal coalescence in chloride systems has been reported by other workers.
P. M. J. Gray reported on the metallothermic reduction of cerous chloride with lithium metal:
This reaction was carried out in a manner very similar to that for the reduction of the trifluoride but was not nearly so successful. In every run a reaction took place satisfactorily but the metal produced would not coalesce and remained finely dispersed throughout the reaction cake. The failure of the metal globules to coalesce is almost certainly due to the presence of oxide or oxychloride which forms an infusible skin on the surface of the metal.
Table 6 compares the apparent current efficiencies of three cerium electrowinning runs. The short duration of the runs, the presence of old electrolyte, lack of information as to the exact valence of the cerium in the compound being electrolyzed, and other cell conditions prevented calculation of actual current efficiencies. For example, in run CE-30 the bath was kept molten by external heating with alternating current, whereas in runs CE-41 and CE-42 direct current was used both for electrolytic and thermal energy.
Apparent current efficiencies, calculated on the basis of total direct current and a valence of 4 for cerium, although useful in comparing one electrolytic run with another, should not be regarded as absolute values.
Average and maximum CO2 and CO in the cell-box atmospheres for runs CE- 30, 41, and 42 show no correlation with the amounts of carbon and oxygen in the cerium metal nodules. (See table 7.)
The present work indicates that the 73 percent CeF3, 15 percent LiF, 12 percent BaF2 mixture is satisfactory for the solvent phase of the electrolyte. With longer runs, however, loss by volatilization may necessitate finding a substitute for LiF.
Considerable cerium and misch metals are prepared commercially by electrowinning from cerous chloride in the air, using the frozen electrolyte as a cell cover. At present, cerous chloride is reported to be cheaper than ceric oxide because it is an intermediate product from the processing of monazite concentrates for thorium. Future electrowinning investigations using cerous chloride as the source of high-purity cerium metal, under controlled atmospheric and temperature conditions similar to those described in this paper, might be worthwhile.
Although there are many important points of similarity between the basic electrochemistry and mechanics of transfer of ions in the commercial aluminum cell and the electrowinning of cerium at the Reno laboratories, two essential differences are apparent to the laboratory investigator.
The first difference is the smaller working volume of the 6-in-diameter pilot cerium electrowinning cell as compared to that of the commercial aluminum cell. The most direct and harmful effect of the smaller volume is that it forces a technique of intermittent operation of the cell; that is, the time of electrolysis is limited by the volume under the electrodes and above the protecting layer of frozen electrolyte available for collecting cerium metal. For example, in test run CE-41, 716 grams of cerium metal accumulated in an electrolysis of 3.65 hours, and the approximate space occupied by 120 cerium nodules prepared in this period was 50 cubic inches. The total space available for collecting cerium nodules was 67 cubic inches. Possibly the nodules did not coalesce into a single mass under the bottom ends of the vertical cathodes because the temperature gradient was too steep in this area due to the restricted space and the need for maintaining a frozen protective layer of the electrolyte above the graphite bottom of the cell. (See fig. 1, p. 15.) The temperature in the end of the graphite anode in test run CE-41 ranged from 752° to 822° C. and averaged 785° C. during electrolysis. The temperatures were higher on the cathode surfaces than at the anode, as the respective current densities were 10.0 and 4.1 a. per sq. cm. Anode density is based on the size and shape of anodes before electrolysis. In test run CE-41 the temperature in the graphite cell bottom was 630° to 728° C. and averaged 675° C. The coalescence of the molten metal droplets at the same temperature depends upon surface tension, viscosity, and relative density of metal and electrolyte. The molten cerium metal deposits on the round cathode and slips down to the end of the rod, where it sinks into the electrolyte and solidifies into irregular, rounded nodules approximately ¼ by ½ by ¾ inch in size.
The other important difference in the 6-inch-diameter, graphite, pilot cerium cell and the commercial aluminum cell is in the geometry of the electrode arrangement and its effect on the cathode products.
The aluminum cell has vertical carbon anodes and a molten horizontal aluminum cathode. This arrangement of electrodes gives the anode gas more freedom to exit without contacting the molten aluminum, compared with the 6-inch-diameter cerium pilot cell where both the anodes and cathodes are parallel and vertical. Moreover, as soon as a molten droplet of cerium metal leaves the surface of the cathode, it sinks into a zone of decreasing temperatures and is prevented from coalescing with the nearest neighboring nodule. The molten aluminum cathode layer, being horizontal and above the melting point of aluminum, does not have this temperature gradient problem.
Other electrodes and electrode arrangements for cerium electrowinning cells are to be investigated in the future.
Electrical-conductivity data are the result of the initial 6 months’ work in the Physics of Metals unit and show only the progress made to establish the relationship between the several grades of cerium metal and electrical conductivity.
Future Work Plans
The next major phase in the development of a process for electrowinning cerium ingot from its oxide is to improve the molten, steady-state or equilibrium conditions. Although the operations in the next phase will be semi-continuous, the volume of bath will be large enough to permit runs five times as long as any reported in this paper. Thus, it will be possible to predict what to expect in a continuous cell, and the investigations will yield data that can be translated into conditions approaching commercial possibilities. Present plans are to use a cell 12 inches in diameter by 12 inches deep. The cell will be enclosed in a larger cell box, designed for closer control of the composition, temperature, and pressure of the atmosphere in the box and for closer control of the temperature of the cell and molten bath. The cell will be equipped with a continuous, mechanical oxide feeder. This feeder will be an important step forward, because a uniform and controlled rate of feeding the oxide is essential to maintaining equilibrium in the molten bath.
Eventually, a method of casting ingot cerium in an inert-atmosphere chamber connected to this cell box will be developed, thus permitting more continuous cell runs.
The longer runs and steadier conditions in the 12-inch cell and associated equipment will provide better control for selective refining of electrolyte and will yield data on aspects of the process that could not be developed with the smaller cell, such as watt-hours per pound of ingot, current efficiencies, and current densities. Furthermore, improved coalescence of the metal might be achieved in the larger bath unit. This improvement, together with the larger quantities of metal product, would result in metal that is more homogeneous and uniform in quality. Confidence that conditions necessary for coalescence can be obtained was encouraged when about 50-percent coalescence of 1,055 grams of metal product was achieved in one run, No. CE-46.
Several months will elapse before the projected cell is in operation. Meanwhile, plans are being made to investigate certain other process variables. Among these variables are oxide feed rate, amount of carbon dioxide that can be tolerated in the box atmosphere, higher purity carbon anodes, and helium cooling of cell accessories.
Ultimately, it is desired to determine the maximum possible rate of feed. Subsequent operations have shown that feed rates higher than those used in the runs described in this report are feasible. A rate of 5.2 grams of ceric oxide per minute at an amperage of 276 has been used successfully for a 3-½- hour test run.
Future electrolyte studies will continue the efforts to measure molten-state properties of the currently used electrolyte, such as solubilities of CeO2 and will continue research on the nature of the phenomena that occur during electrolysis of ceric oxide.
Electrical-conductivity studies on higher purity cerium ingot will be continued to establish more precise data on the relation of purity to specific contamination and to develop data on the true resistivity of higher purity cerium. Investigations will be initiated on the coalescence of individual nodules of cerium using ultrasonics. Methods will be developed for helium cooling the electrowinning cell boxes and accessories.