Table of Contents
A simplified, routine method for determining lithium in lithium-bearing rock is presented. The sample is decomposed by repeated treatments with nitric, hydrofluoric, and perchloric acids. Since this procedure does not appreciably dissolve beryl, interference by beryllium (the principal interfering cation) is minimized for most samples. The salts are brought into solution with water and enough citric acid-ammonium citrate buffer is added to make the final volumetric dilution 0.20 M with respect to citrate. The addition of citrate greatly reduces anion interference in the hydrogen-oxygen flame, slightly increases the intensity of the lithium 671 millimicron emission line, and reduces the effect of most of the interfering cations to the extent that analytical separations are not necessary. The accuracy of results of single determinations of lithium in a pegmatite containing spodumene, lithium-bearing mica, and beryl are approximately equal to that obtained by other flame methods.
Introduction
This investigation was undertaken in Bureau of Mines laboratories to develop a reliable, rapid, routine analytical method for determining lithium in lithium-bearing minerals. The requirements of the procedure were that the method should be applicable to the analysis of a variety of lithium-bearing ores and test products, and the results would be reasonably accurate for materials containing 0.05 to 8 percent Li2O.
Two methods of analysis considered were: (1) Spectrographic, and (2) solution-flame photometric. A short laboratory investigation of spark and arc spectrographic methods was made but the results appeared too erratic for routine lithium analyses in the expected range, whereas flame photometry showed more promise, and attention was focused on developing a procedure that would satisfy the requirements.
Solution-flame photometric methods of analysis for the alkali metals are widely used, but they are known to be sensitive to interference from certain cations and anions. The elimination of interfering ions by chemical separations was recommended by Broderick and Zack and Ellestad and Horstman. Berry, Chappell, and Barnes, Redding, Phillips, and Hiester, Strange, and Williams and Adams found that relatively accurate lithium analyses could be obtained by adding interfering ions to the reference standards in the quantities that occur in the solutions being analyzed. In the present study, it was anticipated that this method would be too tedious, because the quantities of the elements in the unknowns would be variable and would require chemical analysis to determine the proper quantities to be added to the reference standards.
This report discusses the adverse influence of certain ions on the solution-flame photometric determination of lithium and presents a simple means of minimizing their effect.
Apparatus
The instrument used for the investigation was a Beckman, Model DU, spectrophotometer having a 9200 flame attachment. The unit was operated by a selector switch set at 0.1, the sensitivity control near maximum, and the wave length dial setting at 671 millimicrons. Oxygen and hydrogen pressures were varied to obtain the maximum intensity of the lithium flame with each new burner used. The slit width of the unit was adjusted to bring the instrument readings to predetermined values. The detector used was a red-sensitive cesium cell supplied by the manufacturer. Readout in all cases was percent transmission; the term “relative intensity” is used throughout this report to refer specifically to the instrument reading for the emission of the 671 millimicron line of lithium.
Experimental
Parks, Johnson, and Lykken found that the indiscriminate use of acids would result in appreciable errors in the flame photometric determination of alkali metals. Some investigators tried to circumvent this difficulty by adding a fixed amount of acid to both the unknown and reference standards. However, none of the investigators presented the direction and magnitude of errors produced by the hydrogen ion over an extended pH range.
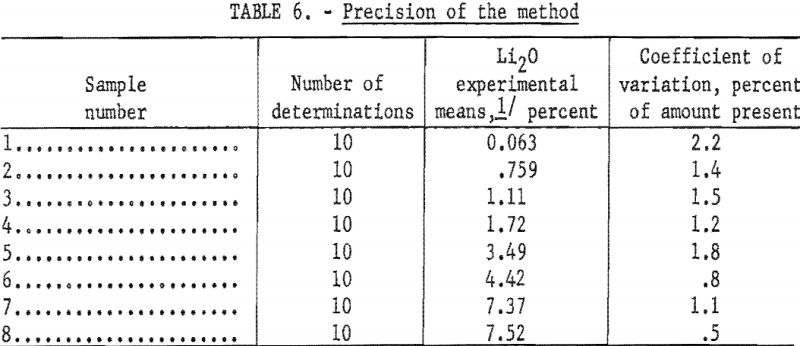
To study the effect of hydrogen ion concentration on the intensity of the lithium flame, solutions, prepared from reprecipitated LiF and containing 30 parts per million (p.p.m.) Li, were adjusted to various pH values over the range of 0.56 to 11.40 by adding either hydrochloric acid (HCl) or barium hydroxide Ba(OH)2. Ba++ ions in low concentrations have little effect on the relative intensity of the lithium flame. The instrument was set at 85.0 relative intensity for the solution at pH 4.02. Results are given in table 1 and are shown graphically in figure 1.
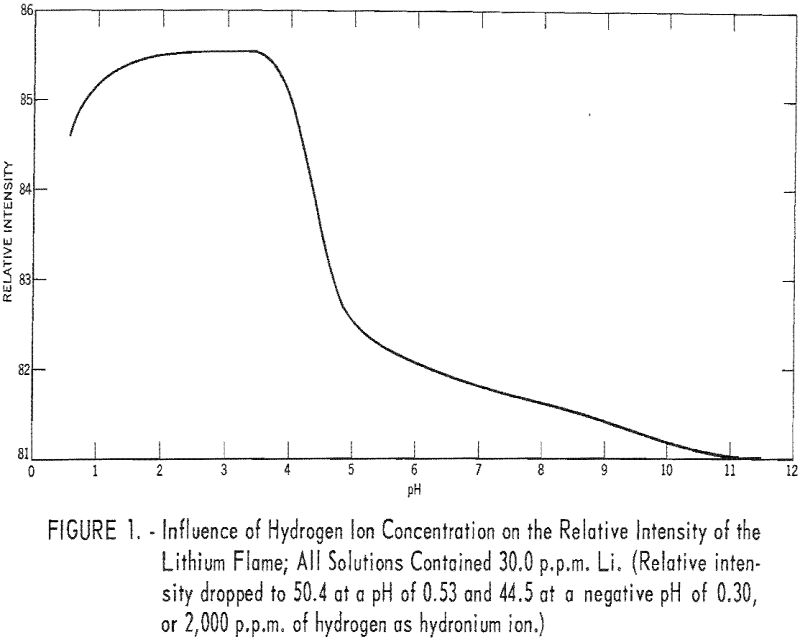
Between pH 1.42 and 3.68 the relative intensity of the lithium flame was maximum and constant. Between pH 0.78 and 4.02 the deviations were small and for most applications could be ignored.
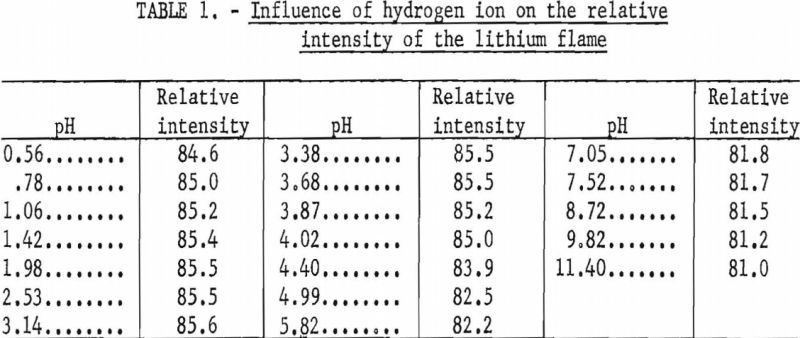
The addition of an organic buffer, citric acid and ammonium citrate, for pH control resulted in an appreciable reduction in bias introduced by most cations and all common anions. However, because organic materials are known to enhance the flame emission of some ions, the amount and direction of this enhancement was determined over a buffer range of 0.02 to 0.40 M. The results of tests made with solutions containing 10, 30, and 50 p.p.m. Li were averaged and are shown graphically in figure 2. Changes in the relative intensity of the lithium flame are referred to readings taken at the 0.20 M citrate buffer level. The pH of the solutions was in the range of 3.5 to 3.7.
The effect of citrate ion, Al+++ and Fe+++ on the lithium flame is shown in table 2. All data are referred to the relative flame intensity of corresponding concentrations of lithium and buffer without the interfering ions.
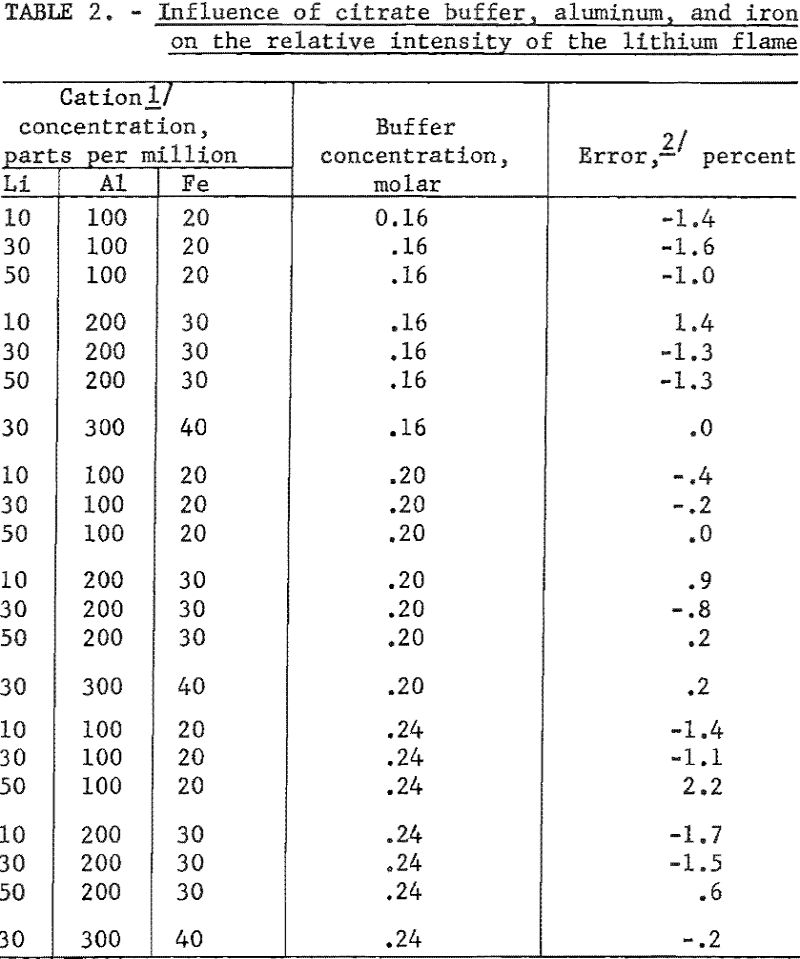
The effect of other single added ions on the intensity of the lithium flame is presented in table 3. The influence of combinations of ions on the flame intensity are given in table 4. Data in both tables are taken from Benson. The concentrations of the lithium and citrate ions are 30 p.p.m. and 0.20 M, respectively, for data in table 3. Readings referred to are for a solution of 0.20 M with respect to citrate and containing 30 p.p.m. Li as the perchlorate. No pH data were taken.

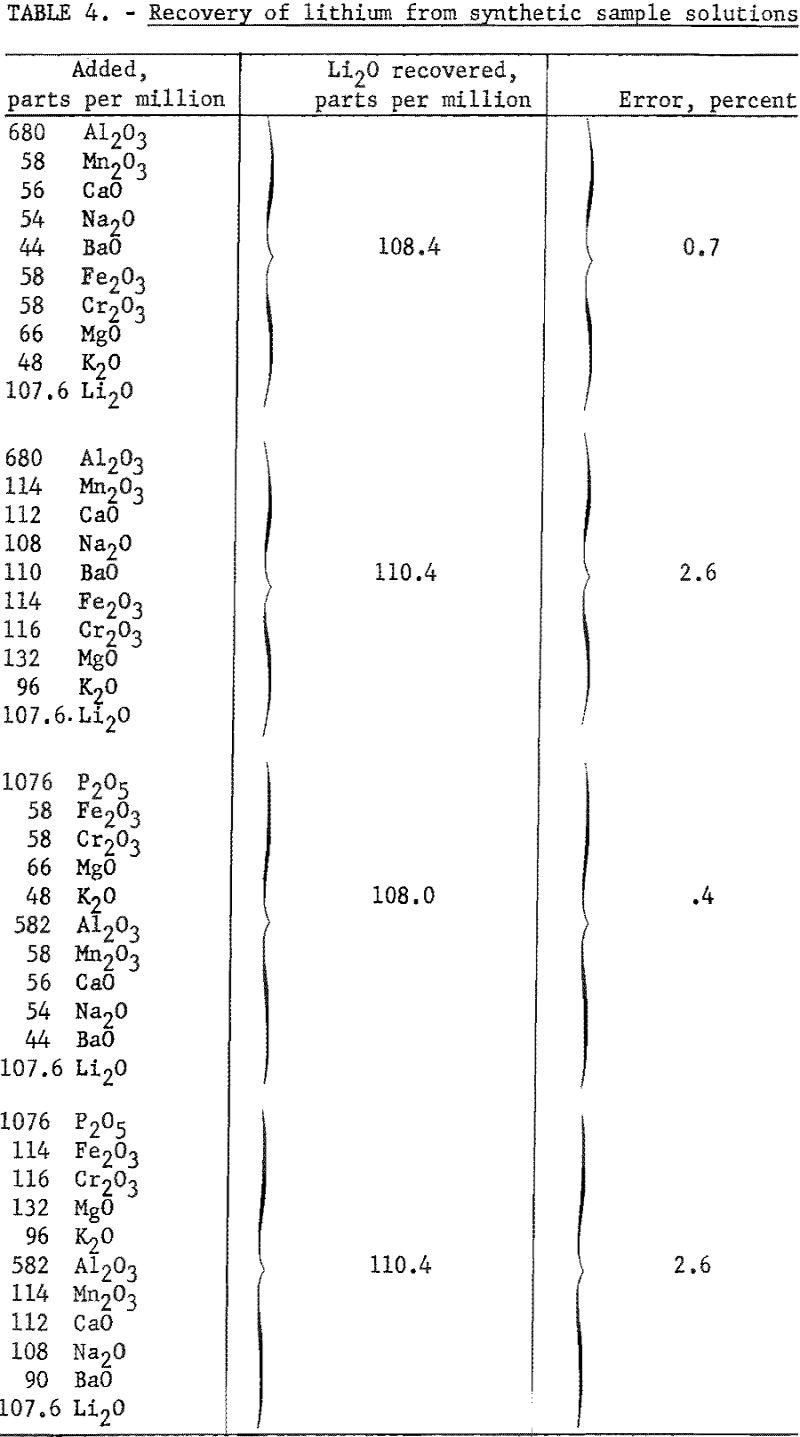
According to these results, a simple routine flame photometric method for determining lithium in minerals seems to be feasible. Except for beryllium, the interfering ions, usually encountered in significant quantities in the analysis of pegmatitic materials were rendered substantially inactive by the citrate buffer. By using the method developed, one set of standards, prepared from pure lithium salts, may be used for reference in analyzing samples of widely varied composition without the necessity of removing the normally occurring interfering ions, or correcting for sample matrix. Beryllium, the principal interfering cation (encountered as beryl in pegmatitic materials), is not appreciably brought into solution by the sample decomposition method used in this investigation. Acid-soluble beryllium minerals, if present in sufficient quantity, would introduce error by beryllium interference.
The details of the procedure are given in following sections.
Preparation of 2 M Citrate Buffer Stock Solution
Weigh 210 grams of citric acid monohydrate and 226 grams of diammonium citrate into a 1-liter beaker. Add 600 to 800 milliliters (ml.) water and boil. Cool, dilute to 1 liter with sterile water and store in a sterile glass bottle under refrigeration. The solution must be discarded and replaced at the first visual indication of mold formation.
In normal analyses, the pH of sample solutions to which this buffer has been added fall between 2.9 and 3.6, depending upon the technique used in dissolving the sample.
Standardization
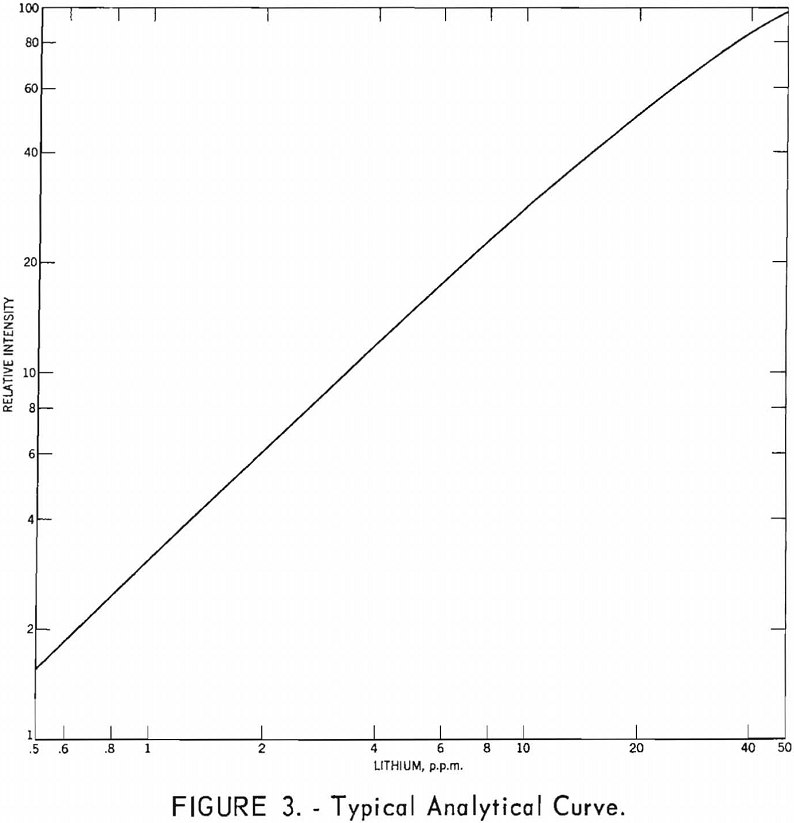
The standard lithium solutions and curves for calibrating the spectrophotometer are prepared as follows: Weigh 3.7378 grams of recrystallized LiF (dried at 110° C.) into a platinum dish, and add 5 ml. nitric acid and 6 to 8 ml, perchloric acid. Evaporate the solution nearly to dryness as described in the preceding section. Dissolve salts in warm water and transfer to a 1-liter volumetric flask. The concentration of this solution is 1,000 p.p.m. Li. Prepare 100-ml. standard solutions, each containing 10.0 ml. of 2 M citrate buffer, at concentrations of 50, 45, 40, 35, 30, 27.5, 25, 22.5, 20, 17.5, 15, 12.5, 10, 8, 6, 5, 4, 3, 2, 1.5, 1, 0.75, and 0.5 p.p.m. Li. Plot instrument readings against concentrations on log-log paper and draw a smooth curve through the points. Determine the Li concentration of the sample solutions from this curve and report as percent Li2O.
After the shape and slope of the analytical curve have been determined, the use of three standard solutions is sufficient for daily calibration. One standard solution at a concentration of 50 p.p.m. Li is used to set the instrument sensitivity at 100 percent transmission.
A typical analytical curve is shown in figure 3.
Photometric Analytical Procedure
For best results the sample to be analyzed should be finer than 200 mesh (Tyler scale). Weigh a 0.25- or 0.5-gram portion, depending on the lithium content of the sample, into a platinum dish. The instrumental range of analysis is 0.05 to 5.4 percent Li2O with a 0.5-gram sample.
To the dish and contents add 5 ml. perchloric acid, 5 ml. concentrated nitric acid, and 10 ml. hydrofluoric acid. Evaporate slowly (1 hour) to the first heavy fumes. Cool, add 5 to 10 ml. hydrofluoric acid and evaporate as above. Repeat the hydrofluoric acid treatment for a total of not more than five evaporations. The final evaporation should be continued until nearly all of the perchloric acid is fumed off. The operation must be watched closely at this point. If evaporation proceeds too far or too fast, an insoluble hydrated oxide of iron may be formed, in which instance the solution must be filtered to prevent clogging of the flame burner. Filtration can be avoided if the evaporation is stopped when the residue appears to be nearly dry but before the fuming has subsided. When a yellow-brown film begins to form, the evaporation must be stopped immediately.
Cool the dish and contents, add water, warm until the salts are in solu-tion, and then transfer to a 250-ml. volumetric flask. Add 25.0 ml. of buffer solution, adjust the temperature and volume, and mix. Allow the insoluble materials, principally beryl, to settle. Use a portion of the clear solution for analysis. Read percent transmission and determine the lithium content from the standard curve.
Most spodumene samples are not completely decomposed by the above procedure. Experience has shown that additional treatments of such samples with hydrofluoric acid have little effect on the insoluble lithium-bearing residue. Because hydrofluoric acid attacks beryl slowly, an excessive amount of this acid should not be used. Instead, the lithium error, amounting to 0.8 to 1.0 percent of the total amount present, can be established for any spodumene by the flame analysis of an ammonium hydroxide filtrate from a pyrosulfate fusion of five or more replicate residues. The sample analyses then can be corrected by this amount.
Interferences
The flame interference caused by has been discussed. Strontium ion gives a weak band interference and should be removed when present. A sulfate separation is satisfactory if only a very small excess of SO4 is added to the solutions.
Accuracy and Precision
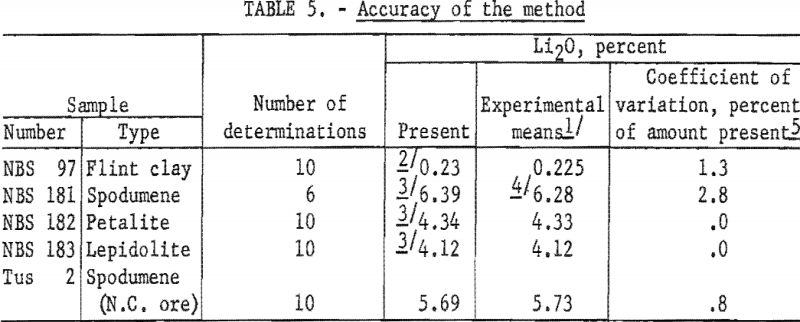
Results of analyses of four National Bureau of Standards (NBS) samples and a North Carolina spodumene ore are presented in table 5.
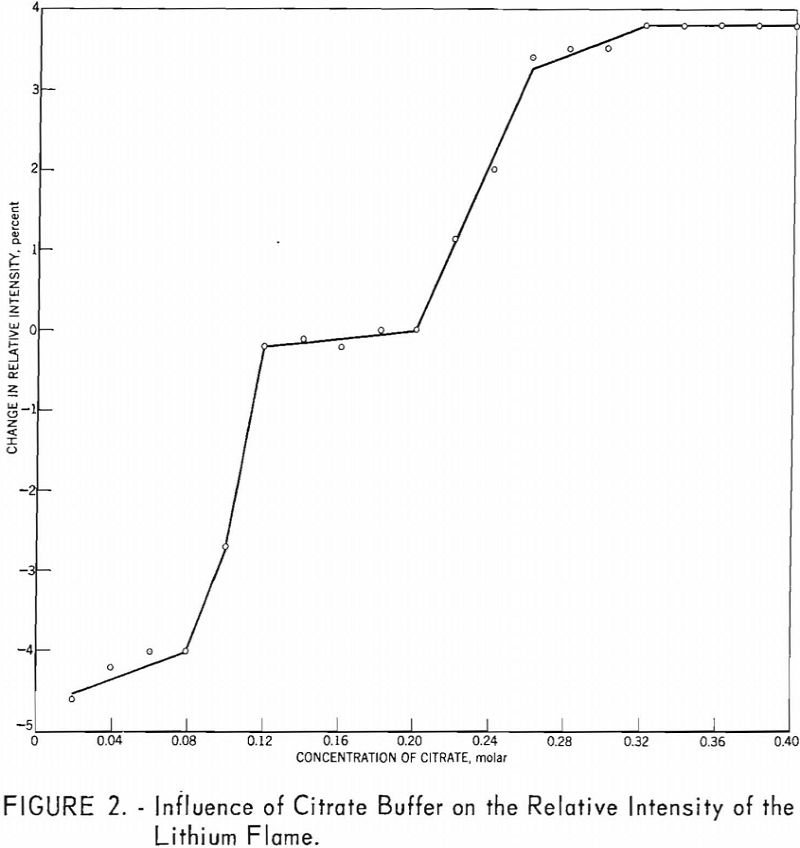
The precision of the method for eight miscellaneous spodumene ore samples in the range of 0.063 to 7.52 percent Li2O is given in table 6.